本篇介紹如何在 Ubuntu Linux 中安裝與使用 QIIME2 微生物組分析流程軟體。
從 Miniconda 官方網站下載安裝檔:
# 下載 Miniconda
wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
執行下載的 Miniconda 安裝檔:
# 安裝 Miniconda
sh Miniconda3-latest-Linux-x86_64.sh
Miniconda 預設會安裝在 $HOME/miniconda3 目錄之下,安裝好之後,將 conda 套件更新至最新版本:
# 更新 conda 套件
conda update conda
並安裝 wget 套件:
# 安裝 wget 套件
conda install wget
Anaconda 也可以直接安裝 Qiime2 套件,所以如果是習慣使用 Anaconda 的人也可以改用 Anaconda 來安裝。
從 Qiime2 官方網站下載 conda 用的套件定義檔:
# 下載 Qiime2 套件定義檔
wget https://data.qiime2.org/distro/core/qiime2-2019.10-py36-linux-conda.yml
根據 Qiime2 套件定義檔建立新的 conda 環境:
# 根據 Qiime2 套件定義檔建立新的 conda 環境,並將其命名為 qiime2-2019.10 conda env create -n qiime2-2019.10 --file qiime2-2019.10-py36-linux-conda.yml
建立好 conda 環境之後,就完成 Qiime2 的安裝了,此時可將沒用的 Qiime2 套件定義檔刪除:
# 刪除 Qiime2 套件定義檔
rm qiime2-2019.10-py36-linux-conda.yml
若要使用 Qiime2,要先載入 qiime2-2019.10 這個剛建立好的 conda 環境:
# 載入 qiime2-2019.10 conda 環境
conda activate qiime2-2019.10
在 qiime2-2019.10 這個 conda 環境中,就可以使用 Qiime2 的指令或 Python 函式庫了。
# 顯示 Qiime2 版本 qiime --version
q2cli version 2019.10.0 Run `qiime info` for more version details.
若要卸載目前所處的 conda 環境,可以執行:
# 卸載目前 conda 環境
conda deactivate
如果在一般的環境中,想要以 qiime2-2019.10 這個 conda 環境來執行特定指令,可以使用 conda run:
# 在 qiime2-2019.10 conda 環境中執行指令 conda run -n qiime2-2019.10 qiime --version
q2cli version 2019.10.0 Run `qiime info` for more version details.
除了 conda run 之外,亦可考慮 conda-wrappers 方式。
如果要移除 qiime2-2019.10 這個 conda 環境,可以執行:
# 移除 qiime2-2019.10 conda 環境 conda env remove -n qiime2-2019.10
Qiime2 官方文件中的 Moving Pictures 實作範例中,分析了人類微生物組的樣本資料,此資料來自於兩個人、四個部位、五個時間點,而第一時間點有使用抗生素,這些資料是在 Illumina HiSeq 上面以地球微生物組計劃(Earth Microbiome Project,簡稱 EMP) hypervariable region 4 (V4) 16S rRNA sequencing protocol 所定序出來的。
首先建立並進入操作範例用的目錄:
# 建立並進入範例目錄 mkdir qiime2-moving-pictures-tutorial cd qiime2-moving-pictures-tutorial
載入 qiime2-2019.10 這個 conda 環境:
# 載入 qiime2-2019.10 conda 環境
conda activate qiime2-2019.10
下載實驗設計相關的後設資料(metadata),這個檔案是一個以 tab 分隔欄位的表格檔案,裡面有樣本資料的基本資訊,包含樣本 ID、barcode、取樣部位、取樣時間、取樣個體、有無使用抗生素等:
# 下載實驗設計相關的後設資料 wget -O sample-metadata.tsv https://data.qiime2.org/2019.10/tutorials/moving-pictures/sample_metadata.tsv
若要將自己的資料整理成 Qiime2 可用的格式,可參考 Qiime2 的後設資料格式說明文件。
下載本分析範例要用的測序片段(reads)資料:
# 建立放置資料的目錄 mkdir emp-single-end-sequences # 下載 barcodes 資料 wget -O emp-single-end-sequences/barcodes.fastq.gz https://data.qiime2.org/2019.10/tutorials/moving-pictures/emp-single-end-sequences/barcodes.fastq.gz # 下載 sequences 資料 wget -O emp-single-end-sequences/sequences.fastq.gz https://data.qiime2.org/2019.10/tutorials/moving-pictures/emp-single-end-sequences/sequences.fastq.gz
通常定序的原始檔案格式都是 FASTQ 格式,若要將資料交給 Qiime2 來處理,必須先將資料轉為 Qiime2 artifact 的格式,而在轉換時需要指定資料的 semantic type,以這個範例來說,semantic type 就是 EMPSingleEndSequences。
# 將原始資料轉為 Qiime2 artifact 格式 qiime tools import --type EMPSingleEndSequences --input-path emp-single-end-sequences --output-path emp-single-end-sequences.qza
EMPSingleEndSequences 就是代表 multiplexed 的序列資料(EMP 標準混樣單端數據),尚未拆分樣本,所以裡面會包含 sequences.fastq.gz 與 barcodes.fastq.gz 兩個檔案。關於更詳細的匯入資料說明,可參考 Qiime2 匯入資料的文件。
在拆分樣本(demultiplex sequences)時我們需要知道哪一個 barcode 對應到哪一個樣本,這個資訊就包含在剛剛上面的後設資料 sample-metadata.tsv 表格中,我們可以使用 demux emp-single 指令來進行樣本的拆分:
# 按照 Barcodes 拆分樣本(Demultiplexing Sequences) qiime demux emp-single --i-seqs emp-single-end-sequences.qza --m-barcodes-file sample-metadata.tsv --m-barcodes-column barcode-sequence --o-per-sample-sequences demux.qza --o-error-correction-details demux-details.qza
其中 --i-seqs 參數用於指定輸入的序列資料,--m-barcodes-file 與 --m-barcodes-column 參數用於指定 barcode 的來源檔案與欄位名稱,--o-per-sample-sequences 用來指定拆分後的序列資料輸出檔案,--o-error-correction-details 則是 Golay 錯誤修正的細節(可用 metadata tabulate 指令來查看內容)。
關於 demux emp-single 指令拆分樣本的使用說明,可參考 Qiime2 官方文件。
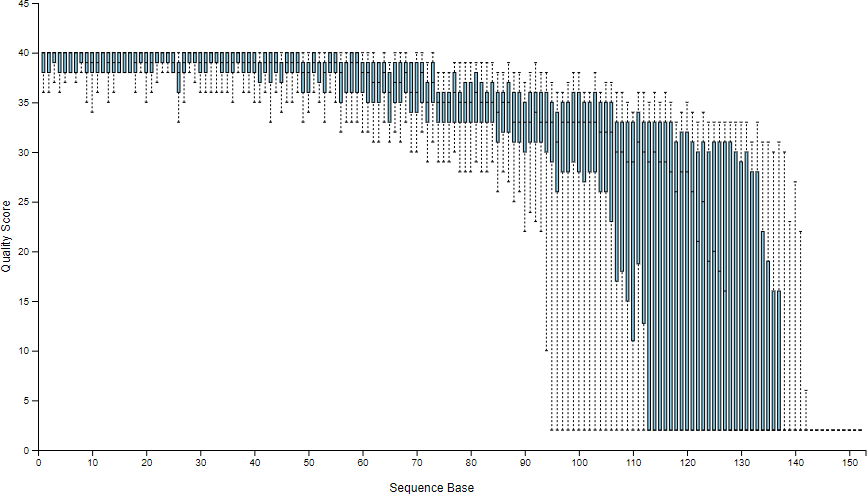
樣本拆分之後,可以顯示簡單的樣本統計資訊,讓我們得知每一個樣本中有多少序列資料,以及序列中各位置的品質分佈資訊:
# 產生樣本拆分後的統計資訊 qiime demux summarize --i-data demux.qza --o-visualization demux.qzv
這裡會產生一個 demux.qzv 視覺化呈現的檔案,裡面包含各種統計資訊,*.qzv 檔案可以用 tools view 指令來查看(需要 XWindow 顯示環境):
# 查看統計結果
qiime tools view demux.qzv
如果沒有 XWindow 的環境可以顯示,亦可將 *.qzv 檔案上傳至 Qiime2 View 網站以網頁方式查看。這個範例可以直接查看官方提供的 demux.qzv 檔案。
序列品質分佈圖中顯示了序列中各位置的品質分佈資訊(Phred quality score),以此例來說左側的序列品質非常好,越往右側則越差。
序列品質管制(quality control,即去噪)方法有很多種,這裡介紹 DADA2 與 Deblur 這兩種方法的使用方式,在實際應用上只需要挑一種方法來使用即可,而不管用哪一種方法,所產生的資料都是以下兩種 Qiime2 artifact:
FeatureTable[Frequency]FeatureData[Sequence]DADA2 是一個偵測與修正 Illumina 擴增子序列資料的處理流程(pipeline),此處理流程會過濾掉 phiX 測序片段(commonly present in marker gene Illumina sequence data),同時也會過濾掉嵌合體(Chimeras)。
DADA2 方法可以透過 dada2 denoise-single 指令來執行,此指令使用時需要指定兩個參數:
--p-trim-left mm 代表每一條序列左側要截去的序列長度。--p-trunc-len nn 代表每一條序列要取用的序列長度。這兩個參數可讓使用去除序列中品質較差的部分,只留下品質好的部分進行分析,而 m 與 n 這兩個值就要依據上面所產生的序列品質分佈圖來決定,以此例來說,序列左側的品質都非常好,所以左側並不需要截去,n 就設定為 0 即可,而右側大約在 120 的位置左右品質就往下掉很多,所以我們就取用 120 以前的序列資料就好,m 就設定為 120。
# DADA2 pipeline:偵測與修正 Illumina 擴增子序列資料 qiime dada2 denoise-single --i-demultiplexed-seqs demux.qza --p-trim-left 0 --p-trunc-len 120 --o-representative-sequences rep-seqs-dada2.qza --o-table table-dada2.qza --o-denoising-stats stats-dada2.qza
產生視覺化資料:
# 產生視覺化資料 qiime metadata tabulate --m-input-file stats-dada2.qza --o-visualization stats-dada2.qzv
如果後續的分析想要採用 DADA2 的輸出資料,則將 DADA2 的輸出檔案重新命名:
# 後續採用 DADA2 的輸出資料繼續分析
mv rep-seqs-dada2.qza rep-seqs.qza
mv table-dada2.qza table.qza
Deblur uses sequence error profiles to associate erroneous sequence reads with the true biological sequence from which they are derived, resulting in high quality sequence variant data.
Deblur 方法有兩個步驟,第一步根據 quality scores 初始化品質篩選流程,這個方法就是 Bokulich et al. (2013) 品質篩選方法的實做版本。
# Deblur 方法 qiime quality-filter q-score --i-demux demux.qza --o-filtered-sequences demux-filtered.qza --o-filter-stats demux-filter-stats.qza
第二步執行 deblur denoise-16S,這裡的 --p-trim-length n 需要指定一個裁剪長度 n,Deblur 開發者建議將 n 設定在 quality score 中位數降至過低的位置,以此例來說適合的 n 大約在 115 到 130 之間,這個值是主觀決定的,在做橫跨多組序列分析的後設分析(meta-analysis)時,也有可能會使用超出建議範圍的 n 值,在這種後設分析中通常會使用統一的 n 值,避免不同研究方法所產生偏差(bias)。由於在上面的 DADA2 方法中我們採用了長度為 120 的序列,而這個值也在建議的範圍內,所以這裡就將 n 設定為 120。
# Deblur 方法 qiime deblur denoise-16S --i-demultiplexed-seqs demux-filtered.qza --p-trim-length 120 --o-representative-sequences rep-seqs-deblur.qza --o-table table-deblur.qza --p-sample-stats --o-stats deblur-stats.qza
以上兩個指令會各產生一個包含統計資訊的檔案,若需要查看檔案內容,可以使用 metadata tabulate 與 deblur visualize-stats 將其轉換為視覺化的 *.qzv 檔案:
# 產生視覺化資料 qiime metadata tabulate --m-input-file demux-filter-stats.qza --o-visualization demux-filter-stats.qzv qiime deblur visualize-stats --i-deblur-stats deblur-stats.qza --o-visualization deblur-stats.qzv
如果後續的分析想要採用 Deblur 的輸出資料,則將 Deblur 的輸出檔案重新命名:
# 後續採用 DADA2 的輸出資料繼續分析
mv rep-seqs-deblur.qza rep-seqs.qza
mv table-deblur.qza table.qza
在後續的分析中,我們會採用 DADA2 的結果。
經過序列的品質篩選程序之後,可以執行以下兩個指令產生視覺化的摘要資訊,feature-table summarize 指令可以產生序列數量分佈的圖表,而 feature-table tabulate-seqs 則可產生 feature identifier 與序列的對應表,同時提供以 BLAST 比對 NCBI nt 資料庫的超連結。
# 產生特徵表視覺化資料 qiime feature-table summarize --i-table table.qza --o-visualization table.qzv --m-sample-metadata-file sample-metadata.tsv qiime feature-table tabulate-seqs --i-data rep-seqs.qza --o-visualization rep-seqs.qzv
Qiime2 支援多種系統發育多樣性度量函數(metrics),包含 Faith’s Phylogenetic Diversity、Weighted UniFrac 與 Unweighted UniFrac,除了各樣本的特徵(features)數量(FeatureTable[Frequency] Qiime2 artifact)之外,這些度量函數還需要各特徵之間有根的系統演化樹(rooted phylogenetic tree),而系統演化樹可以使用 phylogeny align-to-tree-mafft-fasttree 指令來產生,其輸出的 semantic type 為 Phylogeny[Rooted]。
此這一條處理流程會先以 mafft 對 FeatureData[Sequence] 中的序列進行多重序列比對(multiple sequence alignment),產生 FeatureData[AlignedSequence] Qiime2 artifact。
Next, the pipeline masks (or filters) the alignment to remove positions that are highly variable. These positions are generally considered to add noise to a resulting phylogenetic tree.
接著套用 FastTree 依據 masked alignment 產生系統演化樹,FastTree 所產生的是無根的系統演化樹(unrooted phylogenetic tree),所以最後一步要在這一棵樹當中選兩個距離最遠的節點,將樹根設定在中間。
# 產生有根的系統演化樹 qiime phylogeny align-to-tree-mafft-fasttree --i-sequences rep-seqs.qza --o-alignment aligned-rep-seqs.qza --o-masked-alignment masked-aligned-rep-seqs.qza --o-tree unrooted-tree.qza --o-rooted-tree rooted-tree.qza
Qiime2 的 q2-diversity plugin 提供了計算 alpha 與 beta 多樣性度量、相關的統計檢定與視覺化呈現功能,一開始我們先使用 diversity core-metrics-phylogenetic 指令將 FeatureTable[Frequency] 稀疏化(rarefy)至指定的深度(也就是二次抽樣成統一的樣本數),然後計算各種 alpha 與 beta 多樣性度量,並以 Emperor 產生 principle coordinates analysis(PCoA)分析圖。預設會產生的多樣性度量如下:
這裡的 --p-sampling-depth 需要指定一個抽樣深度,這個數值就代表重新抽樣(也就是 rarefaction)的深度,由於大部分的多樣性分析對於不同樣本有不同的抽樣深度非常敏感,所以這裡將每一個樣本都重新隨機抽樣成指定的深度。
舉例來說,假設 --p-sampling-depth 指定為 500,則每一個樣本都會以取後不放回的方式,將每一個樣本重新抽樣成深度為 500 的樣本,如果有樣本原本的深度就不足 500,則該樣本就會在多樣性分析時被丟棄。
選擇抽樣深度需要一些技巧,建立參考一下 table.qzv 的資訊,盡量選擇較大的抽樣深度以保留較多的序列,但是同時也要避免丟棄過多的樣本,在兩者之間取一個適合的值。
# 計算 Alpha 與 Beta 多樣性度量 qiime diversity core-metrics-phylogenetic --i-phylogeny rooted-tree.qza --i-table table.qza --p-sampling-depth 1103 --m-metadata-file sample-metadata.tsv --output-dir core-metrics-results
這裡我們將取樣深度設定為 L3S313 這一組樣本的序列數 1103,這樣就會將所有的樣本深度重新抽樣成 1103,而有三組樣本的原始深度低於這個值,就會被丟棄。此處被丟棄的三組樣本都是屬於 right palm 的樣本,捨棄的樣本不平均並不是很理想的做法,但為了整體保留較多的序列資料,只能以這種折衷的方式處理。
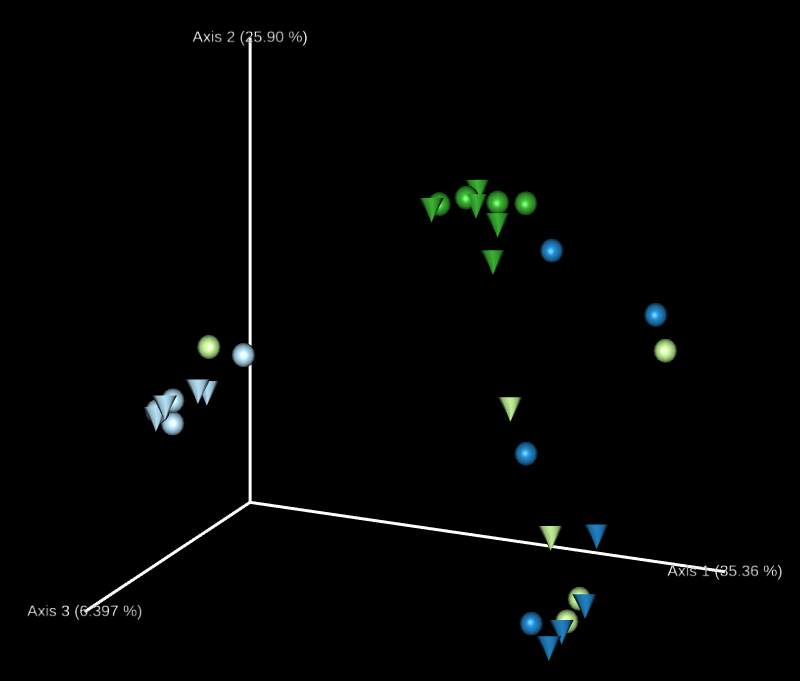
這裡會自動產生許多 3D 的 principal coordinates 圖形,我們可以透過 Emperor 網頁自由調整圖形上面的顏色、大小、樣式如何對應各種資料,呈現想要的結果。
算出各種多樣性度量之後,即可開始探索微生物的結構,這個資訊儲存在一開始下載的後設資料 sample-metadata.tsv 中,首先對類別型欄位與 alpha 多樣性度量進行檢定,這裡選用 Faith’s Phylogenetic Diversity 與 Pielou’s Evenness index 兩種。
# 檢定 Faith's Phylogenetic Diversity 在不同群體的差異 qiime diversity alpha-group-significance --i-alpha-diversity core-metrics-results/faith_pd_vector.qza --m-metadata-file sample-metadata.tsv --o-visualization core-metrics-results/faith-pd-group-significance.qzv # 檢定 Pielou's Evenness index 在不同群體的差異 qiime diversity alpha-group-significance --i-alpha-diversity core-metrics-results/evenness_vector.qza --m-metadata-file sample-metadata.tsv --o-visualization core-metrics-results/evenness-group-significance.qzv
此處所採用的檢定方法為無母數方法中的 Kruskal-Wallis 檢定,對所有群體以及各群體兩兩配對進行中位數差異檢定。
在這個資料集中,沒有連續性的後設資料欄位會跟 alpha 多樣性度量有關係,所以我們在這裡沒有進行連續性變量的檢定,如果有需要連續性變量的檢定,可以使用 diversity alpha-correlation 指令。
接下來我們要使用 diversity beta-group-significance 指令以 排列型多變量變異數分析(PERMANOVA)方法分析樣本結構,看看群體內的樣本間距離是否比群體外的樣本間距離更短,若加上 --p-pairwise 可以同時進行兩兩群體配對的檢定,由於這是一種根據排列(permutation)的無母數檢定,執行時需要一些時間,因此我們只針對有興趣的欄位進行檢定,採用 Unweighted UniFrac distance 對 body-site 與 subject 兩種分群進行檢定。
# PERMANOVA 檢定 qiime diversity beta-group-significance --i-distance-matrix core-metrics-results/unweighted_unifrac_distance_matrix.qza --m-metadata-file sample-metadata.tsv --m-metadata-column body-site --o-visualization core-metrics-results/unweighted-unifrac-body-site-significance.qzv --p-pairwise # PERMANOVA 檢定 qiime diversity beta-group-significance --i-distance-matrix core-metrics-results/unweighted_unifrac_distance_matrix.qza --m-metadata-file sample-metadata.tsv --m-metadata-column subject --o-visualization core-metrics-results/unweighted-unifrac-subject-group-significance.qzv --p-pairwise
此處同樣沒有連續型的資料會與樣本結構有關,若有需要連續型的檢定,可以使用 metadata distance-matrix 配合 diversity mantel 與 diversity bioenv 指令。
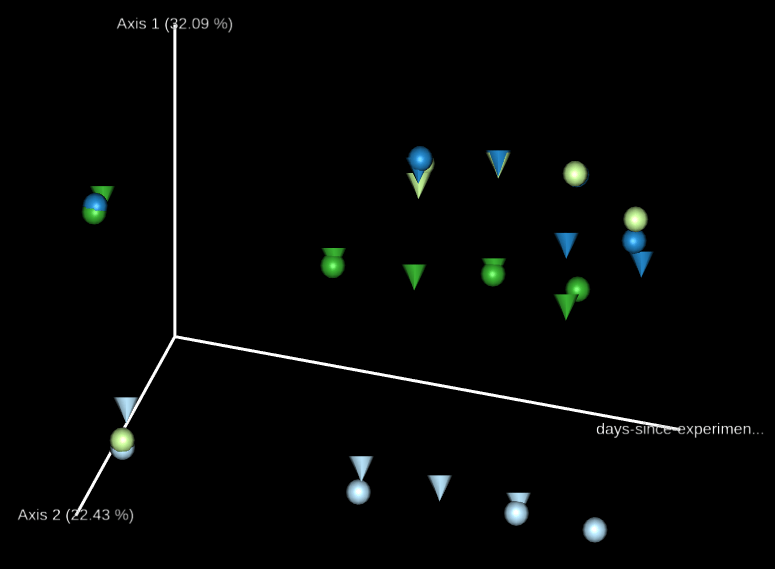
排序分析(ordination analysis)是探討生物群聚與環境之間關係常用的多變量統計方法,我們可以使用 Emperor 這個工具來探索 3D 的 PCoA 圖,core-metrics-phylogenetic 已經有產生了一些基本的圖形,如果想要自行指定特別的座標軸,可以使用 --p-custom-axes 參數,這個很適合用來呈現時間序列的資料。
我們可以直接拿 core-metrics-phylogeny 所產生的 PCoA 結果來產生新的 Emperor 視覺化輸出,這裡我們分別使用 Unweighted UniFrac 與 Bray-Curtis 的前兩個 principal coordinates 搭配實驗經過天數的資料,畫出 3D 的圖形,觀察兩個 principal coordinates 隨著時間的變化。
# 畫出 principal coordinates 與實驗經過天數的關係(Unweighted UniFrac) qiime emperor plot --i-pcoa core-metrics-results/unweighted_unifrac_pcoa_results.qza --m-metadata-file sample-metadata.tsv --p-custom-axes days-since-experiment-start --o-visualization core-metrics-results/unweighted-unifrac-emperor-days-since-experiment-start.qzv # 畫出 principal coordinates 與實驗經過天數的關係(Bray-Curtis) qiime emperor plot --i-pcoa core-metrics-results/bray_curtis_pcoa_results.qza --m-metadata-file sample-metadata.tsv --p-custom-axes days-since-experiment-start --o-visualization core-metrics-results/bray-curtis-emperor-days-since-experiment-start.qzv
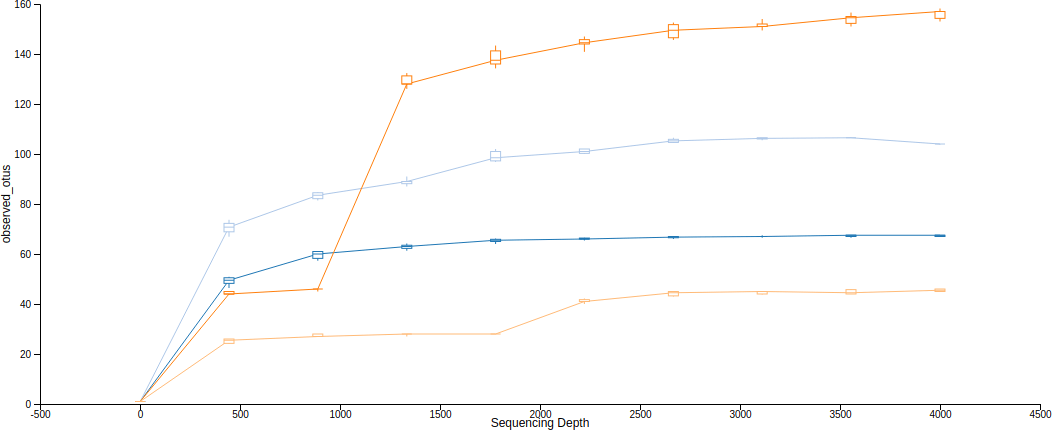
Alpha 稀疏曲線(rarefaction curves)就是呈現 alpha 多樣性與取樣深度的一種關係曲線,在不同的取樣深度下計算出 alpha 多樣性度量,觀察 alpha 多樣性度量(豐富度,richness)是否趨於穩定,從這種圖形中可以判斷取樣深度是否充足。
Qiime2 可以使用 diversity alpha-rarefaction 來畫出 alpha 稀疏曲線,--p-min-depth 與 --p-max-depth 可以指定取樣深度的下限(預設為 1)與上限,在這個區間中取 10 個不同的取樣深度,分別對每一個樣本計算稀疏化的 alpha 多樣性度量,重複操作產生一個預估的區間(--p-iterations 可控制重複次數),最後加上 --m-metadata-file 參數指定後設資料(metadata)來源,這樣就可以在視覺化的界面中選擇不同的分群顯示方式。
# Alpha 稀疏曲線(rarefaction curves) qiime diversity alpha-rarefaction --i-table table.qza --i-phylogeny rooted-tree.qza --p-max-depth 4000 --m-metadata-file sample-metadata.tsv --o-visualization alpha-rarefaction.qzv
這裡產生的圖有上下兩張,第一張是 alpha 稀疏曲線,用來判定豐富度是否已經完全被觀測到(取樣深度是否足夠),如果圖形上的曲線已經趨於平緩,表示絕大部分的特徵都已經被觀測到,增加取樣深度大概也不太會觀測到新的特徵,反之若是曲線末端還是呈現持續上升的趨勢,那就表示可能還有許多特徵尚未被發現,目前的取樣深度尚且不足以表現出整體的樣貌,除此之外,定序錯誤率過高也會造成曲線末端持續上升。
The bottom plot in this visualization is important when grouping samples by metadata. It illustrates the number of samples that remain in each group when the feature table is rarefied to each sampling depth. If a given sampling depth d is larger than the total frequency of a sample s (i.e., the number of sequences that were obtained for sample s), it is not possible to compute the diversity metric for sample s at sampling depth d. If many of the samples in a group have lower total frequencies than d, the average diversity presented for that group at d in the top plot will be unreliable because it will have been computed on relatively few samples. When grouping samples by metadata, it is therefore essential to look at the bottom plot to ensure that the data presented in the top plot is reliable.
這我們要搭配後設資料(metadata)來分析樣本的物種組成,第一步是將 FeatureData[Sequence] 序列資料標上物種的註釋,我們將使用預先訓練好的 Naive Bayes 分類器與 q2-feature-classifier plugin 來處理這個工作,這個分類器是從 Greengenes 13_8 99% OTUs 上訓練出來的,其中序列被修減至僅包含來自於 16S 區域的 250 個鹼基,該 16S 區域在該分析中採用 V4 區域的 515F/806R 引物擴增並定序。我們將把這個分類器應用於我們的序列資料中,這樣即可生成從序列對應到物種的視覺化圖形。
# 下載 Naive Bayes 分類器 wget -O "gg-13-8-99-515-806-nb-classifier.qza" "https://data.qiime2.org/2019.10/common/gg-13-8-99-515-806-nb-classifier.qza" # 進行物種分類 qiime feature-classifier classify-sklearn --i-classifier gg-13-8-99-515-806-nb-classifier.qza --i-reads rep-seqs.qza --o-classification taxonomy.qza # 產生視覺化表格 qiime metadata tabulate --m-input-file taxonomy.qza --o-visualization taxonomy.qzv
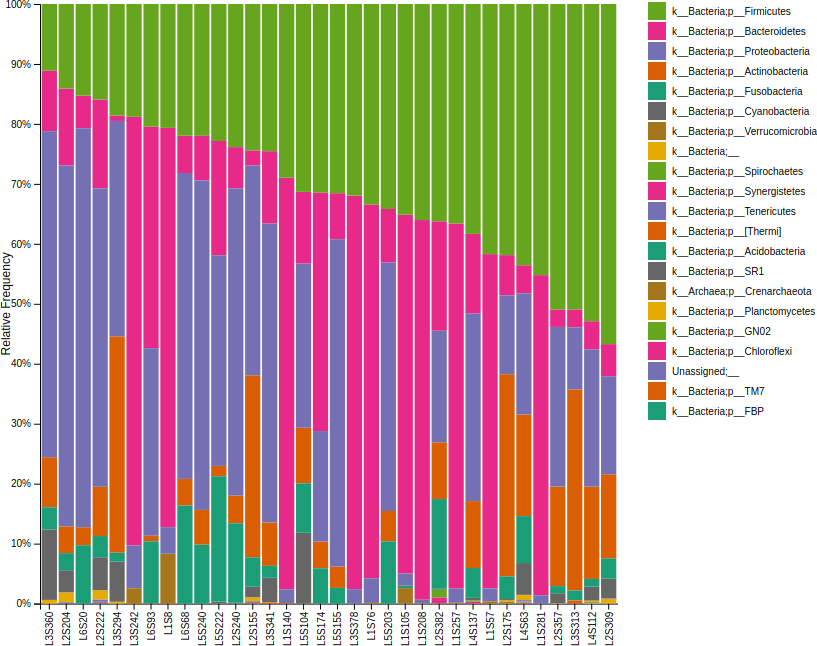
接著產生物種組成長條圖:
# 產生物種組成長條圖 qiime taxa barplot --i-table table.qza --i-taxonomy taxonomy.qza --m-metadata-file sample-metadata.tsv --o-visualization taxa-bar-plots.qzv
差異豐富度分析這裡採用 ANCOM 方法(q2-composition plugin),使用前要注意這個方法的假設與限制,建議先看一下 ANCOM 原始的論文。
q2-gneiss 與 q2-composition 兩個 plugin 可以使用,這裡我們採用 q2-composition,若想要了解 q2-gneiss 的用法,可以參考另外一篇 gneiss 的教學文章。ANCOM 假設不同群體內的特徵差異少於 25%,若不同群組間的特徵差異高於這個比例,則不適合使用 ANCOM 方法(型一誤差與型二誤差皆可能提高),在此例中我們預期身體的不同部位會有較高的差異,所以我們只針對腸道(gut)的樣本進行分析,以 ANCOM 判斷在兩個個體的腸道樣本中,是否存在豐富度差異(?)。
首先產生腸道(gut)的特徵表(關於篩選資料的方法請參考Filtering data 的教學文件):
# 產生腸道(gut)的特徵表 qiime feature-table filter-samples --i-table table.qza --m-metadata-file sample-metadata.tsv --p-where "[body-site]='gut'" --o-filtered-table gut-table.qza
ANCOM 需要 FeatureTable[Composition] 這種資料,其來自於每一個樣本的特徵數量,但是數量不可以是 0。使用 composition add-pseudocount 可以將 FeatureTable[Frequency] 轉為 ANCOM 所需要的 FeatureTable[Composition](加入假的數量):
# 製作 ANCOM 需要的 FeatureTable[Composition](避免數量為零) qiime composition add-pseudocount --i-table gut-table.qza --o-composition-table comp-gut-table.qza
We can then run ANCOM on the subject column to determine what features differ in abundance across the gut samples of the two subjects.
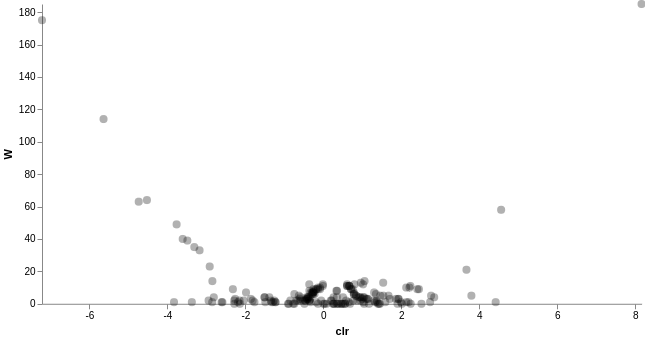
以 ANCOM 方法檢定兩個個體腸道中的樣本是否存在豐富度差異:
# ANCOM 差異豐富度分析 qiime composition ancom --i-table comp-gut-table.qza --m-metadata-file sample-metadata.tsv --m-metadata-column subject --o-visualization ancom-subject.qzv
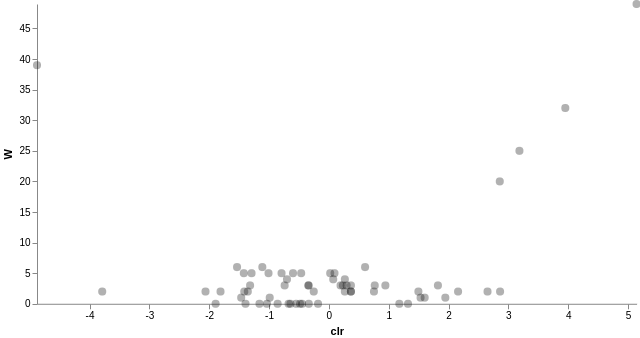
我們也可以針對指定的物種分類層級進行差異豐富度分析,只要產含有物種分類資訊的 FeatureTable[Composition] 表格,在重新執行一次 ANCOM 差異豐富度分析即可:
# 加入物種分類資訊 qiime taxa collapse --i-table gut-table.qza --i-taxonomy taxonomy.qza --p-level 6 --o-collapsed-table gut-table-l6.qza # 製作 ANCOM 需要的 FeatureTable[Composition] qiime composition add-pseudocount --i-table gut-table-l6.qza --o-composition-table comp-gut-table-l6.qza # ANCOM 差異豐富度分析 qiime composition ancom --i-table comp-gut-table-l6.qza --m-metadata-file sample-metadata.tsv --m-metadata-column subject --o-visualization l6-ancom-subject.qzv
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}